A mutation in SCN11A eliminates pain sensation
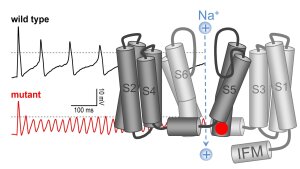
A mutation in SCN11A causes a gain-of-function of the voltage-gated sodium channel NaV1.9 and leads to suppression of the electrical signaling in nociceptive neurons.
Illustration: FSU BiophysikThe sensation of pain is unpleasant but it protects the body from serious injury. In collaboration with the group of Dr. Ingo KurthExterner Link (Institute of Human Genetics, University Hospital Jena) we characterized a heterozygous genetic defect in SCN11A that causes the congenital inability to experience pain in affected humans. Knockin mice carrying the orthologous mutation showed reduced sensitivity to pain and self-inflicted tissue lesions, recapitulating aspects of the human phenotype. SCN11A encodes the voltage-gated sodium channel NaV1.9, which is highly expressed in nociceptive dorsal root ganglia neurons, the key relay stations for the transmission of sensory information from the periphery to the brain. Mutant NaV1.9 channels displayed gain-of-function properties characterized by excessive activity at resting voltages. In this novel channelopathy, hyperactive NaV1.9 channels result in a seemingly counterintuitive suppression of electrical signaling in DRG neurons. Specific activation of NaV1.9 channels may thus constitute an alternative way to modulate pain perception.